Background
Suggested Reading
- Lehninger Chapter 8.1
- Wilson and Walker 4.1 - 4.2 (the properties of nucleic acids)
- Wilson and Walker 4.7.1 (Isolation of DNA)
- Wilson and Walker 4.7.3 (Kit-based extraction of nucleic acids)
Lecture Video
A Brief History of DNA Isolation
Due to its unique chemical properties, nucleic acid polymers, DNA (and RNA) are relatively easy to separate from other components of the cell. In fact, the methods are simple enough that long before scientists knew much about DNA they recruited wailers to isolate gallons of thick DNA paste from whale tissue for research. The procedure you are about to follow has many more chemical components than the procedure you followed to isolate protein. Before I explain the function of each component, I think it is interesting to learn that nobody actually set-out to develop a method for purifying DNA this way. Instead, it was the other way around. In 1869, a young German scientist named Friedrich Miescher precipitated DNA as he was cleaning used surgical bandages with salt, soap, ethanol, and water.1 He thought the stuff he isolated was interesting, but he didn't realize that his discovery would launch one of the greatest scientific revolutions of all time a revolution that would change our fundamental understanding of life, lead to unimaginable medical advances, and even empower humans to make synthetic organisms. I wonder how he would react if someone told him that scientists and students would be replicating variations of his procedure thousands (if not millions) of times every day by the 21st century.
 Let me set the scene to give you an idea of the state of biochemical science at that time. In 1867, at age 23, Friedrich Miescher was inspired by an uncle (an anatomical scientist) who explained that the remaining questions concerning the development of tissues could only be solved on the basis of chemistry. In his last year at University of Tübingen, he trained as an organic chemist for one semester under Adolf Strecker, who was the first person to synthesize amino acids. This gave him enough experience to secure himself an apprenticeship with Felix Hoppe-Seyler, a well-known physiological chemist who headed one of the first biochemical laboratories in history. His laboratory was located in the medieval castle that had recently been purchase by University. It was located high above the old town and the surrounding river valleys (see picture above). As a converted castle, Hoppe-Seyler's main laboratory was in a laundry room. When Friedrich Miescher joined, he was put to work in the former kitchen. Hoppe-Seyler was famous at the time for describing the absorption spectrum of the red pigment in blood using a rudimentary spectrophotometer made of lamps and prisms in 1862 (the German mathematician August Beer had published his famous law only ten years prior). Later, in 1864, he crystallized the protein hemoglobin, the substance responsible for that color and demonstrated that it carries oxygen.
Let me set the scene to give you an idea of the state of biochemical science at that time. In 1867, at age 23, Friedrich Miescher was inspired by an uncle (an anatomical scientist) who explained that the remaining questions concerning the development of tissues could only be solved on the basis of chemistry. In his last year at University of Tübingen, he trained as an organic chemist for one semester under Adolf Strecker, who was the first person to synthesize amino acids. This gave him enough experience to secure himself an apprenticeship with Felix Hoppe-Seyler, a well-known physiological chemist who headed one of the first biochemical laboratories in history. His laboratory was located in the medieval castle that had recently been purchase by University. It was located high above the old town and the surrounding river valleys (see picture above). As a converted castle, Hoppe-Seyler's main laboratory was in a laundry room. When Friedrich Miescher joined, he was put to work in the former kitchen. Hoppe-Seyler was famous at the time for describing the absorption spectrum of the red pigment in blood using a rudimentary spectrophotometer made of lamps and prisms in 1862 (the German mathematician August Beer had published his famous law only ten years prior). Later, in 1864, he crystallized the protein hemoglobin, the substance responsible for that color and demonstrated that it carries oxygen.
Hoppe-Seyler put Miescher on the project of classifying the proteins of white blood cells. To obtain white blood cells, Miescher had to wash them off them off of pus-filled bandages (as the new kid, he was given the job no one else wanted to do). Anyway, during his experiments, he serendipitously noticed a substance with unexpected properties. It could be precipitated in ethanol and saline water. Once isolated, it could be precipitated again in acid and re-dissolved in base. Unknowingly, Miescher had, for the first time, isolated DNA. He was intrigued and performed further experiments to discover from where inside the cells this new substance came. He discovered that it came from the nucleus. Miescher named his molecule nuclein. As scientists learned more about it, the name eventually morphed to deoxyribonucleic acid (DNA). Only three years prior to Miescher's discovery, the influential German biologist Ernst Haeckel had proposed that the nucleus contained the factors responsible for the transduction of hereditary traits. Miescher's isolation of DNA launched a new interest in the nucleus. Over the rest of his career, he learned much about the chemical composition of DNA and even proposed the theory that information was encoded in the chirality of the monomers (of course, incorrect, but a good place to start). It wasn't until decades after Mieschers death that scientists finally figured-out why his DNA isolation method actually works. In the following procedure, we'll go through the standard steps of DNA purification and discuss why they work.
Nucleic Acids
 Early tests on DNA found that it was unlike any other cellular material previously discovered. It precipitated in strong acid (indicating a negative charge), had high phosphorous concentration, and came from the nucleus of the cells. This lead to its early name nuclein, as it was believed to be a protein within the nucleus. Today we know that nucleic acids are a completely different class of macromolecule from proteins.
Early tests on DNA found that it was unlike any other cellular material previously discovered. It precipitated in strong acid (indicating a negative charge), had high phosphorous concentration, and came from the nucleus of the cells. This lead to its early name nuclein, as it was believed to be a protein within the nucleus. Today we know that nucleic acids are a completely different class of macromolecule from proteins.
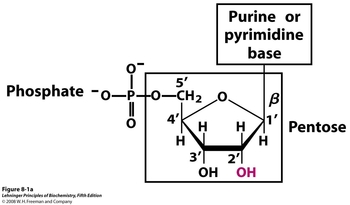
Nucleic acids are comprised of three essential structural components. The core of all nucleic acids is a pentose ring, with the 2' position differentiating RNA from DNA. The 2' hydroxyl in ribonucleic acid is replaced with a 2' Hydrogen in deoxyribonucleic acid. Linked to the 3' position is a phosphate group, which explains both the high phosphate concentration and negative charge observed in their early discovery. Finally, a beta linkage at the 1' position leads to either a purine (adenine, guanine) or pyrimidine (cytosine, thymine/uracil) base. In RNA the thymine base is not used, and is instead replaced with uracil.
(adenine, guanine) or pyrimidine (cytosine, thymine/uracil) base. In RNA the thymine base is not used, and is instead replaced with uracil.
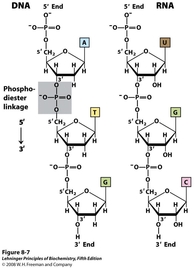
 Strands of nucleic acids are linked by phosphodiester bonds (shaded in the image to the left). The alternating of pentoses and phosphates makes DNA single strands polar. This linkage also gives nucleic acids their directionality, as the 5' terminus of the chain ends in a free phosphate, while the 3' terminus ends in a free hydroxyl group.
Strands of nucleic acids are linked by phosphodiester bonds (shaded in the image to the left). The alternating of pentoses and phosphates makes DNA single strands polar. This linkage also gives nucleic acids their directionality, as the 5' terminus of the chain ends in a free phosphate, while the 3' terminus ends in a free hydroxyl group.
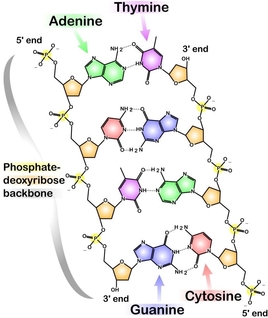
Each strand of DNA binds a complementary strand via hydrogen bonding between complementary bases. Each base has a set of hydrogen bond donors and acceptors so that it will favorably line up with its complementary base. Adenine and Thymine are one complementary set, Cytosine and Guanine are the other. As can be seen in the figure to the right, each base pair is composed of one purine and one pyrimidine. This allows for an optimal spacing (~10 Å) between the two strands. The base pairing in DNA gives the structure its double-helix shape, which allows for the polar pentose-phosphate backbone to be solvent exposed while the hydrophobic bases are buried within the helix core. One additional difference between DNA and RNA is that the 2' hydroxyl in RNA prevents stable formation of a double-stranded complex. Therefore, while DNA occurs naturally as a double-stranded superstructure, RNA is usually single stranded.
DNA in Bacteria
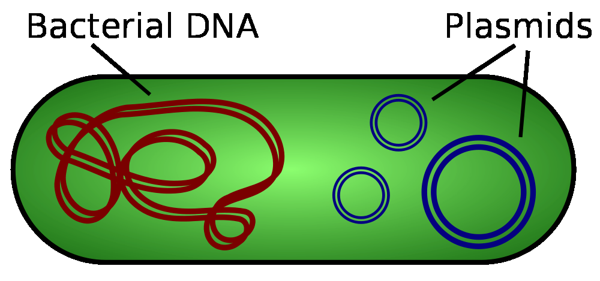 Like all orgnisms, bacteria have genomic DNA that contains all of the information needed for replication. This DNA is a large, covalently closed circular structure containing millions of base pairs. However, certain bacteria also contain smaller sets of circular DNA called plasmids. Plasmids are much smaller than the genomic DNA, usually ranging from 3,000-30,000 base pairs.2 Bacterial plasmids usually encode for specialized protein(s) that allow survival in a given environment. Bacteria growing in saline conditions may contain plasmids which encode for additional ion efflux pumps. Plasmid DNA, although not associated with the host's genomic DNA, is replicated along with the genomic DNA, and is carried over in cell division. Molecular biologists and biochemists have found ways to introduce artificial, non-native, recombinant DNA into bacterial cells in order to produce large numbers of a desired protein. This genetic manipulation using recombinant plasmids forms the basis for our imaginary company in the premise of this experiment.
Like all orgnisms, bacteria have genomic DNA that contains all of the information needed for replication. This DNA is a large, covalently closed circular structure containing millions of base pairs. However, certain bacteria also contain smaller sets of circular DNA called plasmids. Plasmids are much smaller than the genomic DNA, usually ranging from 3,000-30,000 base pairs.2 Bacterial plasmids usually encode for specialized protein(s) that allow survival in a given environment. Bacteria growing in saline conditions may contain plasmids which encode for additional ion efflux pumps. Plasmid DNA, although not associated with the host's genomic DNA, is replicated along with the genomic DNA, and is carried over in cell division. Molecular biologists and biochemists have found ways to introduce artificial, non-native, recombinant DNA into bacterial cells in order to produce large numbers of a desired protein. This genetic manipulation using recombinant plasmids forms the basis for our imaginary company in the premise of this experiment.
Alkaline Lysis Plasmid Isolation
The alkaline lysis plasmid isolation procedure3 is a quick method especially suitable for screening large numbers of E. coli cultures. The bacteria are lysed by treatment with strong base (NaOH) and detergent. The chromosomal DNA denatures and remains associated with the bacterial membrane instead of renaturing. It is then removed by centrifugation. The plasmid DNA remains in the supernatant from which it is then precipitated with ethanol. The procedure is simple and low cost. However, it is more common for laboratories to purchase a commercial kit to carry out this procedure. The majority of plasmid isolation kits available carry-out the exact same process with one major modification, they do not utilize the chloroform-phenol extraction step to remove proteins. Instead, the DNA is precipitated and caught on a short silica column that is washed with an ethanol solution to remove proteins. The DNA is finally washed off the silica column with either buffer or water. This step is convenient in that it avoids the use of toxic chemicals and eliminates the time-consuming drying step of our method. However, it is often noted that the DNA yield from these kits tends to be considerably lower than that of the procedure presented (unpublished observations). Additional details are outlined below.
Resuspending the Cells
 Cells are first resuspended in a Glucose-Tris-EDTA (GTE) buffer.3 The glucose maintains the osmotic pressure of the bacteria, keeping the cells intact until the lysis step. Tris buffer maintains a pH of 8.0, which is near physiological pH to keep the cells viable, but high enough to increase DNA solubility upon lysis. Finally, the EDTA binds all divalent cations (Mg2+, Ca2+) upon lysis. These cations are necessary cofactors for DNA degrading endonucleases.
Cells are first resuspended in a Glucose-Tris-EDTA (GTE) buffer.3 The glucose maintains the osmotic pressure of the bacteria, keeping the cells intact until the lysis step. Tris buffer maintains a pH of 8.0, which is near physiological pH to keep the cells viable, but high enough to increase DNA solubility upon lysis. Finally, the EDTA binds all divalent cations (Mg2+, Ca2+) upon lysis. These cations are necessary cofactors for DNA degrading endonucleases.
Without EDTA, lysing the cell would release endonucleases that would hydrolyze our DNA. EDTA, Ethylenediaminetetraacetic acid (seen on the right), is a compound that chelates large metals by completely saturating their coordination sites. The hexa-dentate nature of EDTA allows it to strip metals from the active sites of endonucleases. Similarly, the dissociation constant is so high that free, divalent metal ions are virtually non-existent, inhibiting the nuclease activity.
Bovine pancreatic RNase A is also added to hydrolyze RNA. This enzyme degrades RNA but uses no metal cofactors. It binds RNA with cationic lysine and arginine. This tough enzyme is active during all steps of the procedure until removed by extraction. Not adding RNase A will preclude accurate measurement of plasmid DNA concentration, as the RNA nucleotides will be co-isolated and UV-active upon characterization.
Lysing the Cells
In order for the DNA to be extracted, the cells must first be lysed. Cell membrane integrity varies depending on the organism. In the mushroom experiments, you used liquid nitrogen to freeze the cells, and a mortar and pestle to physically break down the cell walls. Fortunately, E. coli cells are more easily lysed by simply dissolving the membranes with SDS detergent. The addition of NaOH with SDS increases the pH of the cellular extract, denaturing proteins and DNA alike.2
Denaturing Genomic DNA
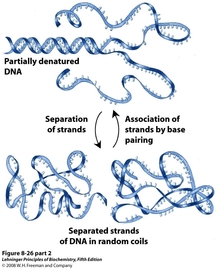 The high pH (>10) of the cellular extract at this point destabilizes the hydrogen bonding interactions that hold the DNA double helix together. This step must be done carefully, for vigorous mixing will shear the genomic DNAinto large pieces that will co-isolate with the plasmid.
The high pH (>10) of the cellular extract at this point destabilizes the hydrogen bonding interactions that hold the DNA double helix together. This step must be done carefully, for vigorous mixing will shear the genomic DNAinto large pieces that will co-isolate with the plasmid.
Once the DNA is separated into single strands, the pH is rapidly dropped to 4.8 by adding a potassium acetate solution. This sudden re-emergence of hydrogen bonding forces the DNA to quickly base pair, leading to aggregation in the large, genomic DNA from the exposed bases that did not correctly pair. The insoluble genomic aggregates precipitate out of solution. The much smaller plasmid DNA, however, is able to correctly base pair upon the change in pH, and remains in solution. The precipitated genomic DNA can now be removed by centrifugation and pipetting off the supernatant.
This step purifies the plasmid DNA from the genomic DNA, but many proteins and small molecules still remain.
Extracting Proteins and Lipids
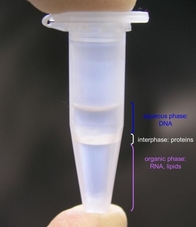 Now that we have plasmid DNA as the only nucleotide macromolecule remaining, we can use the different properties of DNA, proteins, and lipids to our advantage. There are two primary techniques for separating the nucleic acids from other material in a solution, phenol:chloroform extraction (more common for large scale nucleic acid purification) and silica column filtration (more common for small scale work).
Now that we have plasmid DNA as the only nucleotide macromolecule remaining, we can use the different properties of DNA, proteins, and lipids to our advantage. There are two primary techniques for separating the nucleic acids from other material in a solution, phenol:chloroform extraction (more common for large scale nucleic acid purification) and silica column filtration (more common for small scale work).
Phenol:chloroform extraction partitions the solution into two layers with the organic layer on the bottom.2 Phenol further denatures the proteins, making them less soluble in the aqueous phase, and draws their insoluble aggregates to the interface between the phases. The chloroform extracts the lipids, degraded RNA bases, and other organic components of the extract into the organic phase. Often, isoamyl alcohol is also added to the mixture to reduce foaming and emulsion at the interface. The DNA is now isolated in the top, aqueous layer. This layer now must carefully be pipetted off in order to avoid pipetting any of the proteins or phenol, which could complicate the UV characterization (more on this later). Note: Phenol is an inhalation and contact toxin. Pipet it in the fume hood only! Always dispose of the chloroform solution in the ORGANIC HAZARDOUS WASTE.
The method we will use is silica column filtration. At high ionic strength and acidic pH, nucleic acid polymers have a very high affinity for silica gel and everything else in the aqueous solution flows through a silica column. We will use a centrifuge to force the the solution through our column, however vacuum and air pressure are equally effective. Next, a wash solution that is high in both salts and ethanol is used to thoroughly wash the column while retaining the DNA as insoluble on the silica. This is a direct application of Miescher's discovery that DNA is insoluble in high ionic strength ethanol solution. The charges on DNA are less solvated by water when they associate with monovalent cations such as Na+ and K+ instead of divalent Mg2+. This neutralized DNA salt fully precipitates from solution in the presence of ethanol. Finally, the DNA is eluted from the column with either water or Tris-EDTA (TE) buffer. DNA is highly soluble in these solutions, the latter protects DNA from degradation but may be incompatable with some downstream applications.
Solid phase extraction of DNA by silica spin column
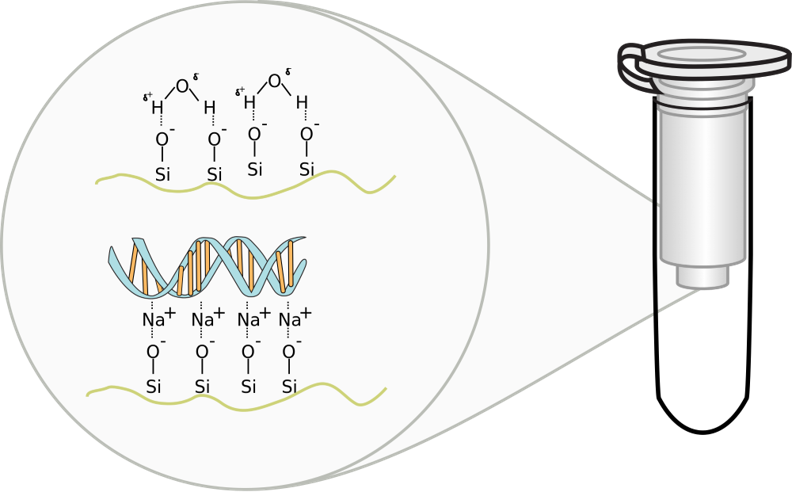
Chaotropic salts, such as sodium iodide, sodium thiocyanate, or sodium perchlorate, cause DNA to bind silica, glass particles, and unicellular algae called diatoms which shield their cell walls with silica (see figure at left). This property was developed to purify nucleic acid using glass powder or silica beads under alkaline conditions in the 1980s. In practice, DNA isolation kits use either guanidinium thiocyanate or guanidinium hydrochloride as the chaotropic agents in the ethanolic wash solution rather than sodium salts due to the empirical observation that protein is more effectively washed away by protein-denaturing guanidinium ions.5 The effect of guanidinium on DNA solubility is comparable to that of sodium.5 In the 1990s, the use of silica gel replaced glass beads when silica spin columns, like the kind we are using, became commerically available.
To bind DNA to silica, the sample must be dissolved in a binding buffer of chaotropic salt. The solution resulting from the neutralization step above works well. This solution is transferred to a spin column and the column is centrifuged. The centrifuge forces the binding solution through a silica gel membrane that is inside the spin column. If the pH and salt concentration of the binding solution are optimal, the nucleic acid will bind to the silica gel membrane as the solution passes through.
Next, the flow-through is removed and a wash buffer is added to the column. The column is put in a centrifuge again, forcing the wash buffer through the membrane. This removes any remaining impurities from the membrane, leaving only the nucleic acid bound to the silica gel.
To elute, the wash buffer is removed and an elution buffer (or simply water) is added to the column. The column is put in a centrifuge again, forcing the elution buffer through the membrane. The elution buffer removes the nucleic acid from the membrane and the nucleic acid is collected from the bottom of the column.
Precipitating and Concentrating Isolated Plasmid
 Oftentimes, the plasmid DNA is precipitated in order to clear it of any phenol that may be left over from the previous step. Pure ethanol is added to lower the polarity of the DNA solution. In this state, the charges on DNA are less solvated by water, and associate with monovalent cations such as Na+ and K+ from whatever buffer was present. The optimal concentration of ethanol for DNA precipitation is 70% v/v. After centrifugation, the ethanol is poured-off leaving the DNA pellet in the tube. Remaining ethanol can be evaporated off to render a dried, DNA salt product.
Oftentimes, the plasmid DNA is precipitated in order to clear it of any phenol that may be left over from the previous step. Pure ethanol is added to lower the polarity of the DNA solution. In this state, the charges on DNA are less solvated by water, and associate with monovalent cations such as Na+ and K+ from whatever buffer was present. The optimal concentration of ethanol for DNA precipitation is 70% v/v. After centrifugation, the ethanol is poured-off leaving the DNA pellet in the tube. Remaining ethanol can be evaporated off to render a dried, DNA salt product.
Many companies that ship purified DNA to customers leave it in this solid, salt form, as it is stable enough to transport at room temperature. However, as we want to characterize the DNA and use it next week for restriction digests, we will not precipitate it.